
 DOI
DOI Code
Code Data
DataQuantum chemistry is pivotal in exploring the behaviors of substances at atomic and molecular levels [1, 2]. Key simulation techniques, such as density functional theory (DFT) [3] and its more efficient variant, density functional tight binding (DFTB) [4], have become essential tools. DFT, based on quantum mechanics principles [5], provides a comprehensive framework for studying the electronic structure of materials, revealing chemical bonding, charge distribution, and other quantum phenomena. However, the computational demands of solving the many-body Schreodinger equation (SE) limit the size of systems that can be feasibly studied [6].
DFTB offers a computationally lighter alternative, allowing the study of larger and more complex systems. While it is less accurate than DFT, DFTB’s efficiency facilitates simulations of extensive molecular systems and materials [7]. However, the use of empirical parameters in DFTB, which may not be universally applicable, presents challenges in ensuring broad reliability across different materials [8].
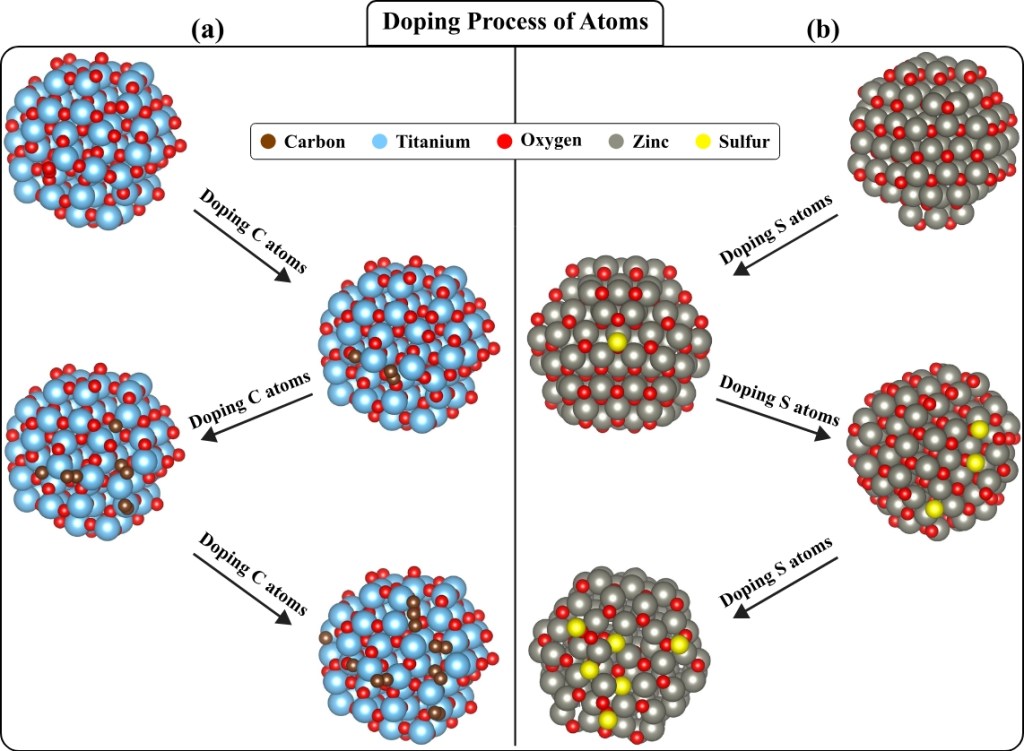
In addressing existing challenges in materials science, this work introduces Pure2DopeNet, a novel multimodal ML approach designed to reduce the computational requirements of material simulations and minimize the need for extensive domain knowledge. Pure2DopeNet combines text- guidance with molecular images of pure compounds to predict multiple physical parameters of doped compounds, focusing on Carbon (C) doped TiO2 [ 45] and Sulfur (S) doped ZnO [46].

Key contributions of this work include: (i) the introduction of a multimodal framework integrating text and image data for compound property prediction; (ii) the ability to predict doped compound properties based on pure form simulations, extending predictive capabilities; (iii) a significant reduction in computational costs compared to traditional methods; and (iv) enhanced accessibility by minimizing the need for complex simulations, broadening its applicability in materials science.
Cite this article (BibTeX):
@article{polat2024multimodal,
author={Polat, Can and Kurban, Mustafa and Kurban, Hasan},
title={Multimodal Neural Network-Based Predictive Modeling of Nanoparticle Properties from Pure Compounds},
journal={Machine Learning: Science and Technology},
year={2024},
}References
[1] Attila Szabo and Neil S Ostlund. Modern quantum chemistry: introduction to advanced electronic structure theory. Courier Corporation, 2012.
[2] Peter W. Atkins and Ronald S. Friedman. Molecular quantum mechanics. Oxford University Press, 2005.
[3] Walter Kohn and Lu J. Sham. Self-consistent equations including exchange and correlation effects. Physical review, 140:A1133, 1965.
[4] G Seifert. Tight-binding density functional theory: an approximate kohn- sham dft scheme. The Journal of Physical Chemistry A, 111(26):5609–5613, 2007.
[5] Jun John Sakurai and Eugene D Commins. Modern quantum mechanics, revised edition. American Association of Physics Teachers, 1995.
[6] Jie Pan. Scaling up system size in materials simulation. Nature Computational Science, 1:95–95, 2021.
[7] Mathias Rapacioli, Aude Simon, Leo Dontot, and Fernand Spiegelman. Extensions of dftb to investigate molecular complexes and clusters. physica status solidi (b), 249(2):245–258, 2012.
[8] Breno RL Galvao, Lu´ıs P Viegas, Dennis R Salahub, and Maicon P Louren¸co. Reliability of semiempirical and dftb methods for the global optimization of the structures of nanoclusters. Journal of Molecular Modeling, 26:1–8, 2020.
[46] Weilai Yu, Jinfeng Zhang, and Tianyou Peng. New insight into the enhanced photocatalytic activity of n-, c-and s-doped zno photocatalysts. Applied Catalysis B: Environmental, 181:220–227, 2016.
